Overview
Autoimmunity occurs when the body's immune system mistakenly attacks healthy tissues, leading to organ damage. Pemphigus vulgaris is a potentially fatal autoimmune disease in which autoantibodies against desmosomal cell adhesion molecules known as desmogleins cause blistering of the skin and mucous membranes. Our laboratory was launched with a mission to understand pathogenic mechanisms in this model organ-specific autoimmune disease, with the goal of using our knowledge to develop precision targeted therapies for pemphigus and other autoantibody-mediated diseases.
Mechanisms of autoimmunity in pemphigus
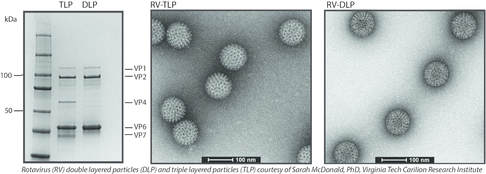
A fundamental question in organ-specific autoimmune disease is why the immune system breaks tolerance against only a limited number of self-antigens. We have cloned B cell repertoires from pemphigus vulgaris patients to understand how they developed desmoglein autoreactivity. We identified shared VH1-46 gene usage in anti-desmoglein 3 B cells from different pemphigus patients and defined acidic amino acid residues that are necessary and sufficient to confer desmoglein 3 autoreactivity. We also identified common as well as divergent features of the B cell response to the self-antigen desmoglein 3 and the rotavirus VP6 antigen, which suggests shared VH gene usage in the immune response to foreign and self antigens as a potential basis for triggering pathologic autoimmune reactions, but that divergent evolution limits the onset of autoimmunity after infection. Further studies have uncovered the developmental pathways and lineage relationships of IgG and IgA B cells in pemphigus, indicating that the IgG4 B cells that are predominant in active disease are clonally distinct from the shared lineages found within the IgG1-IgA1-IgA2 axis.
Regulation of desmosomal cell adhesion in primary human keratinocytes
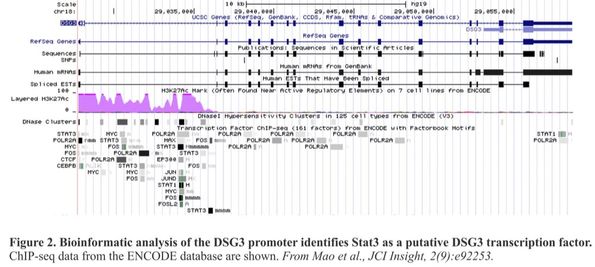
Using the pathologic anti-desmoglein monoclonal antibodies we have isolated from pemphigus patients, our laboratory has also elucidated the cell regulatory pathways in primary human keratinocytes that promote desmosome adhesion. We have shown that the p38 MAPK/MK2 axis governs keratinocyte desmosomal adhesion and that inhibition of this pathway can ameliorate pemphigus skin blistering. We identified STAT3 as a key regulator of desmoglein 3 transcription in keratinocytes, which contributes to the rapid therapeutic effect of corticosteroids in pemphigus and may explain the loss of desmoglein 3 expression in advanced head and neck squamous cell cancers. In addition to providing insight into desmosome biology, these studies have identified adjunctive treatment strategies to block end-organ damage caused by pemphigus autoantibodies.
Precision therapy for B cell-mediated diseases in humans and dogs
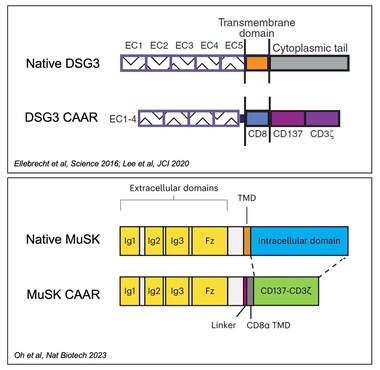
Ultimately, a better understanding of the shared structural elements of autoimmune cell repertoires can lead to better targeted therapies for disease. Current methods for treating autoimmunity require general immune suppression to reduce antibody production, but this approach impairs protective immune responses, which can lead to potentially fatal infections and secondary cancers. We described a novel method for re-engineering chimeric antigen receptor (CAR) T cells, which have led to lasting remissions of B cell-mediated cancers, for targeted immunotherapy of B cell-mediated autoimmune diseases. By using the autoantigen that is the target of autoantibody attack as the extracellular domain of a chimeric autoantibody receptor (CAAR), we can direct a patient’s T cells to specifically seek out and kill autoantibody-expressing B cells, while sparing the healthy immune cells that protect from infection. We published the preclinical proof-of-concept for the CAAR approach using desmoglein 3 CAAR T cells (DSG3-CAART) for mucosal pemphigus vulgaris (Ellebrecht et al, Science 2016). We performed definitive preclinical studies that enabled an Investigational New Drug application for DSG3-CAART (Lee et al, JCI 2020), which is currently being evaluated in a phase 1 clinical trial (NCT04422912). We have extended the CAART concept to the treatment of muscle-specific tyrosine kinase (MuSK) myasthenia gravis (Oh et al, Nature Biotechnol. 2023), which is the subject of a phase 1 clinical trial (NCT05451212), as well as phospholipase A2 receptor membranous nephropathy.
Additionally, we are collaborating with Penn Vet to develop cellular immunotherapies for cancer and autoimmunity in dogs. Following up on the pioneering work of Nicola Mason, BVetMed, PhD, to launch a first-in-canine clinical trial of CD20-targeted CAR T cells in canine B cell lymphoma, we have generated a next-generation anti-CD20 CART that we plan to take forward to canine clinical trials to determine if lasting remission of canine B cell-mediated cancers and autoimmune diseases are possible.
Additionally, we are collaborating with Penn Vet to develop cellular immunotherapies for cancer and autoimmunity in dogs. Following up on the pioneering work of Nicola Mason, BVetMed, PhD, to launch a first-in-canine clinical trial of CD20-targeted CAR T cells in canine B cell lymphoma, we have generated a next-generation anti-CD20 CART that we plan to take forward to canine clinical trials to determine if lasting remission of canine B cell-mediated cancers and autoimmune diseases are possible.
